 |
|
|

The aim of this tutorial is to introduce users to the PLUMED syntax. We will go through the preparation of input files to calculate and print simple collective variables on a pre-calculated trajectory. This tutorial has been prepared by Max Bonomi (adapting a lot of material from other tutorials and Masterclass) for the Master In Silico Drug Design, held at Université Paris Cité on March 6th, 2023.
Once this tutorial is completed, users will be able to:
In this and in the next tutorial, we will use two pieces of software: PLUMED version 2.8.2 and GROMACS version 2019.6. First, we need to install the software on your machine using conda.
First, check if conda is installed on your machine by typing:
conda
In case the command is not found, please follow the instructions below to install conda in the directory /path/to/conda. You can choose it in your home so that you will have write permission to it. If conda is already installed on your machine, please skip the instructions below and move directly to the creation of the conda environment for this tutorial.
# on Linux: wget -c https://repo.continuum.io/miniconda/Miniconda3-latest-Linux-x86_64.sh -O miniconda.sh # on MacOS: # wget -c https://repo.continuum.io/miniconda/Miniconda3-latest-MacOSX-x86_64.sh -O miniconda.sh bash ./miniconda.sh -b -f -p /path/to/conda # export PATH to find the conda executable export PATH="/path/to/conda/bin:$PATH" # initialize bash shell conda init bash
Now we can create a conda environment for the ISDD tutorial using the following commands:
# create environment conda create --name ISDD-tutorial-2023
and activate it with:
# activate environment conda activate ISDD-tutorial-2023
Finally, we can proceed with the installation of the required software:
conda install -c conda-forge plumed conda install --strict-channel-priority -c plumed/label/masterclass -c conda-forge gromacs
The --strict-channel-priority might be necessary in case your conda install is configured to download packages from the bioconda channel. Indeed, bioconda contains a version of GROMACS that is not patched with PLUMED and would thus not work here.
Keep in mind that every time you open a new shell, in order to use PLUMED and GROMACS you need to activate the ISDD-tutorial-2023 environment using the following command:
conda activate ISDD-tutorial-2023
PLUMED is a library that can be incorporated into many MD codes by adding a relatively simple and well documented interface. Once it is incorporated you can use PLUMED to perform a variety of different analyses on-the-fly and to bias the sampling in MD simulations. Additionally, PLUMED can be used as a standalone code for analyzing trajectories. If you are using the code in this way you can run the PLUMED executable by issuing the command:
plumed <instructions>
Let's start by getting a feel for the range of calculations that PLUMED can do. Issue the following command now:
plumed --help
The output of this command is a list of tasks that PLUMED can perform. Among these, there are commands that allow you to patch an MD code, postprocess metadynamics simulations, and build the manual. In this tutorial we will mostly use PLUMED to analyze trajectories. In order to do so, we will learn how to use the driver tool. Let's look at the options of PLUMED driver by issuing the following command:
plumed driver --help
As you can see we can do a number of things with PLUMED driver. For all of these options, however, we are going to need to write a PLUMED input file. The syntax of the PLUMED input file is the same that we will use later to run enhanced sampling simulations during a MD simulation, so all the things that you will learn now will be useful later when you will run PLUMED coupled to GROMACS.
GROMACS is a molecular dynamics package mainly designed for simulations of proteins, lipids, and nucleic acids. More info can be found here.
You can check that GROMACS is properly working by typing:
gmx mdrun -h
If you inspect the long output generated by this command, you will notice a -plumed option, which indicates that GROMACS has properly been compiled with PLUMED. Great, now you are ready to go!
The main goal of PLUMED is to compute collective variables (or CVs), which are complex descriptors of the system that can be used to describe the conformational change of a protein or a chemical reaction. This can be done either on-the-fly during a molecular dynamics simulations or a posteriori on a pre-calculated trajectory using PLUMED as a post-processing tool. In both cases one, you should create an input file with a specific PLUMED syntax. Have a look at the sample input file below:
# Compute distance between atoms 1 and 10. # Atoms are ordered as in the trajectory files and their numbering starts from 1. # The distance is called "d" for future reference. d: DISTANCEATOMS=1,10 # Compute the torsional angle between atoms 1, 10, 20, and 30. # The angle is called "phi1" for future reference. phi1: TORSIONthe pair of atom that we are calculating the distance between.ATOMS=1,10,20,30 # The same CV defined above can be split into multiple lines # The angle is called "phi2" for future reference. phi2: TORSION ...the four atoms involved in the torsional angleATOMS=1,10,20,30 ... # Print "d" on a file named "COLVAR1" every 10 steps. PRINTthe four atoms involved in the torsional angleARG=dthe input for this action is the scalar output from one or more other actions.FILE=COLVAR1the name of the file on which to output these quantitiesSTRIDE=10 # Print "phi1" and "phi2" on another file named "COLVAR2" every 100 steps. PRINTcompulsory keyword ( default=1 ) the frequency with which the quantities of interest should be outputARG=phi1,phi2the input for this action is the scalar output from one or more other actions.FILE=COLVAR2the name of the file on which to output these quantitiesSTRIDE=100compulsory keyword ( default=1 ) the frequency with which the quantities of interest should be output
In the input file above, each line defines a so-called action. In this simple example, actions are used to compute a distance, a dihedral angle, or print some values on a file. Each action supports a number of keywords, whose value is specified. Action names are highlighted in green and, by clicking on them, you can go to the corresponding page in the manual that contains a detailed description of each keyword. Actions that support the keyword STRIDE are those that determine how frequently things are done. Notice that the default value for STRIDE is always 1. In the example above, omitting STRIDE keywords the corresponding COLVAR files would have been written for every frame of the analyzed trajectory. All the other actions in the example above, i.e. DISTANCE and TORSION, do not support the STRIDE keyword and are only calculated when requested. That is, d will be computed every 10 frames, and phi1 and phi2 every 100 frames.
Variables should be given a name (in the example above, d, phi1, and phi2), which is then used to refer to these variables in subsequent actions, such as the PRINT command. A lists of atoms should be provided as comma separated numbers, with no space.
You can find more information on the PLUMED syntax at Getting Started page of the manual. The complete documentation for all the supported collective variables can be found at the Collective Variables page.
By default the PLUMED inputs and outputs quantities in the following units:
If you want to change these units, you can do this using the UNITS keyword.
The TARBALL for this tutorial contains the following files:
GB1_native.pdb: a PDB file with the native structure of the GB1 protein.traj-whole.xtc: a trajectory in xtc format. GB1 has already been made whole by fixing discontinuities due to periodic boundary conditions.traj-broken.xtc: the same trajectory as it was originally produced by GROMACS. Here GB1 is broken by periodic boundary conditions.After dowloading the compressed archive to your local machine, you can unpack it using the following command:
tar xvzf master-ISDD-1.tar.gz
Once unpacked, all the files can be found in the master-ISDD-1 directory. To keep things clean, it is recommended to run each exercise in a separate sub-directory that you can create inside master-ISDD-1.
In this exercise, we will learn how to compute and print collective variables on a pre-calculated MD trajectory. To analyze the trajectories provided here, we will:
plumed.dat);Notice that you can also visualize trajectories with VMD (always a good idea!). For example, the trajectory traj-whole.xtc can be visualized with the command:
vmd GB1_native.pdb traj-whole.xtc
Let's now prepare a PLUMED input file to calculate:
GB1_native.pdb file to retrieve the list of indexes of the CA atoms;R_0 to 8.0 Angstrom (be careful with units!).Below you can find a sample plumed.dat file that you can use as a template. Whenever you see an highlighted FILL string, be careful because this is a string that you must replace.
# Compute gyration radius on CA atoms: r: GYRATIONATOMS=__FILL__ # Compute number of contacts between CA atoms co: COORDINATIONthe group of atoms that you are calculating the Gyration Tensor for.GROUPA=__FILL__First list of atoms.R_0=__FILL__ # Print the two collective variables on COLVAR file every step PRINTcould not find this keywordARG=__FILL__the input for this action is the scalar output from one or more other actions.FILE=COLVARthe name of the file on which to output these quantitiesSTRIDE=__FILL__compulsory keyword ( default=1 ) the frequency with which the quantities of interest should be output
Once your plumed.dat file is complete, you can run the PLUMED driver as follows:
> plumed driver --plumed plumed.dat --mf_xtc traj-broken.xtc
Scroll in your terminal to read the PLUMED log. As you can see, PLUMED gives a lot of feedback about the input that it is reading and the actions that it will execute. Please take your time to inspect the log file and check if PLUMED is actually doing what you intend to do.
The command above will create a COLVAR file like this one:
#! FIELDS time r co 0.000000 2.458704 165.184127 1.000000 2.341932 164.546604 2.000000 2.404708 162.606975 3.000000 2.454297 143.850122 4.000000 2.569342 147.110408 5.000000 2.304027 163.608703
Notice that the first line informs you about the content of each column.
In case you obtain different numbers, check your input, you might have made some mistake!
This file can then be visualized using gnuplot (for more info about this tool, look here):
gnuplot> p "COLVAR" u 1:2, "" u 1:3
Now, look at what happens if you run the exercise twice. The second time, PLUMED will back up the previously produced file so as not to overwrite it. You can also concatenate your files by using the action RESTART at the beginning of your input file.
Finally, let's try to use the same input file with traj-whole.xtc and examine the results. Did you find the same results as with the previous trajectory? Why?
PLUMED provides some shortcuts to select atoms with specific properties. To use this feature, you should specify the MOLINFO action along with a reference PDB file. This command is used to provide information on the molecules that are present in your system.
Let's try to use this functionality to calculate the backbone dihedral angle \( \phi \) (phi) of residue 2 of the GB1 protein. This CV is defined by the action TORSION and a set of 4 atoms. For residue i, the dihedral \( \phi \) is defined by these atoms: C(i-1),N(i),CA(i),C(i) (see Fig. master-ISDD-1-dih-fig).
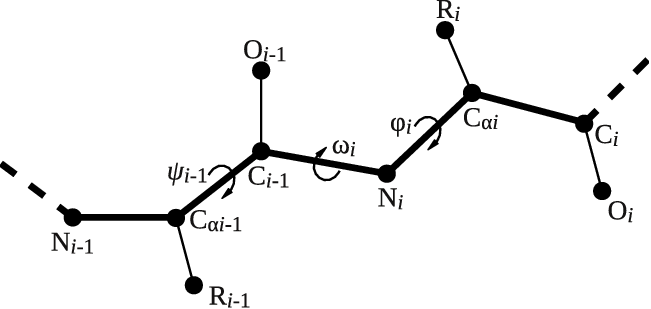
After consulting the manual and inspecting GB1_native.pdb, let's define the dihedral angle \( \phi \) of residue 2 in two different ways:
t1).t2);# Activate MOLINFO functionalities MOLINFOSTRUCTURE=__FILL__ # Define the dihedral phi of residue 2 as an explicit list of 4 atoms t1: TORSIONcompulsory keyword a file in pdb format containing a reference structure.ATOMS=__FILL__ # Define the same dihedral using MOLINFO shortcuts t2: TORSIONthe four atoms involved in the torsional angleATOMS=__FILL__ # Print the two collective variables on COLVAR file every 10 steps PRINTthe four atoms involved in the torsional angleARG=__FILL__the input for this action is the scalar output from one or more other actions.FILE=COLVARthe name of the file on which to output these quantitiesSTRIDE=__FILL__compulsory keyword ( default=1 ) the frequency with which the quantities of interest should be output
After completing the PLUMED input file above, let's use it to analyze the trajectory traj-whole.xtc using the driver tool:
> plumed driver --plumed plumed.dat --mf_xtc traj-whole.xtc
You can use gnuplot to visualize the trajectory of the two CVs calculated with the PLUMED input file above and written in the COLVAR file. Are the two trajectories identical?
In summary, in this tutorial you should have learned how to use PLUMED to:
 1.8.17
1.8.17 
