 |
|
|
| |
|
|
In this tutorial I will show you how you can use PLUMED and metadynamics to study the binding between a ion and a dinucleotide.
Once this tutorial is completed students will
The reference trajectory and other files can be obtained at this path
wget https://github.com/plumed/lugano2019/raw/master/lugano-6b.tgz
This tutorial has been tested on v2.5 but it should also work with other versions of PLUMED.
For this tutorial we will consider a practical application. The system is actually taken from [40]. It is one of the simplest systems studied in that paper, namely a guanine dinucleotide monophosphate binding a Mg ion. This is a very simple binding event, but a very similar procedure might be used to study binding of a ligand on a protein. The reason why this exercise is particularly simple is that instead of the ligand we have a single ion, thus with no internal degree of freedom, and instead of a protein with a complex binding pocket we have a dinucleotide. We are also assuming to know which is the proper binding site, since we can easily guess that the most stable binding will happen on the phosphate.
Since running these simulations on your laptop would take too long, you will start with the output files obtained with a decently long simulation and analyze them.
Before continuing, please read carefully the plumed.dat file that was used to produce the simulation, since there you will find all the explanations about which variables were biased and how.
In case you want to do analysis with python, you can use the included plumed_pandas.py module, which is a preview of a feature that will be available in plumed 2.6. It requires pandas to be installed (use conda install pandas) and allows to extract columns from a COLVAR file by name. It works in this way:
> import plumed_pandas
> import matplotlib.pyplot as plt
> df=plumed_pandas.read_as_pandas("COLVAR")
# shows the head of the file:
> df.head()
# plot distance between Mg and phosphate
> plt.plot(df["dp"][:],".")
# plot coordination number of Mg with water
> plt.plot(df["cn"][:],".")
As the title says, just compute the free-energy landscape as a function of the biased collective variable (namely, distance between the Mg ion and the phosphate and coordination number of the Mg ion with water oxygen atoms). You should just use sum_hills with the usual options. In order to visualize the result with gnuplot you might use something like this:
gnuplot> set pm3d map gnuplot> sp "fes.dat" u 1:2:3
You should obtain a plot similar to this one:

This exercise is optional and is not needed to continue with the next points. However, it is a very good idea to do it in order to have a better understanding of what the system is doing!
Beware that the periodic boundary conditions were broken. You can adjust them using PLUMED with an input like this one (please fill the gaps)
MOLINFOSTRUCTURE=conf.pdb WHOLEMOLECULEScompulsory keyword a file in pdb format containing a reference structure.ENTITY0=@nucleic c: CENTERthe atoms that make up a molecule that you wish to align.ATOMS=@nucleic mg: GROUPthe list of atoms which are involved the virtual atom's definition.ATOMS=__FILL__ # find the serial number of the Mg ion WRAPAROUNDthe numerical indexes for the set of atoms in the group.AROUND=creference atoms.ATOMS=mg # check documentation of WRAPAROUND! # you should also know how many atoms make a water molecule WRAPAROUNDwrapped atoms.AROUND=creference atoms.ATOMS=@waterwrapped atoms.GROUPBY=__FILL__ # dump your trajectory DUMPATOMScompulsory keyword ( default=1 ) group atoms so as not to break moleculesATOMS=@mdatomsthe atom indices whose positions you would like to print out.FILE=whole.xtc # writing all atoms you will be able to reuse the same pdb for opening. # e.g. vmd conf.pdb whole.xtccompulsory keyword file on which to output coordinates; extension is automatically detected
Now reweight your free energy and compute it as a function of:
The free energy as a function of the distance between Mg and geometric center of RNA can be used to identify the bulk region. In order to do so, normalize it adding the correct entropic term \(k_BT\log d^2\), and find a region where the free energy is approximately constant to represent the bulk region. You can for instance use the following commands in gnuplot
gnuplot> p "fes_dc" u 1:2 , "" u 1:($2+2.5*log($1**2))
Below you can find reference results

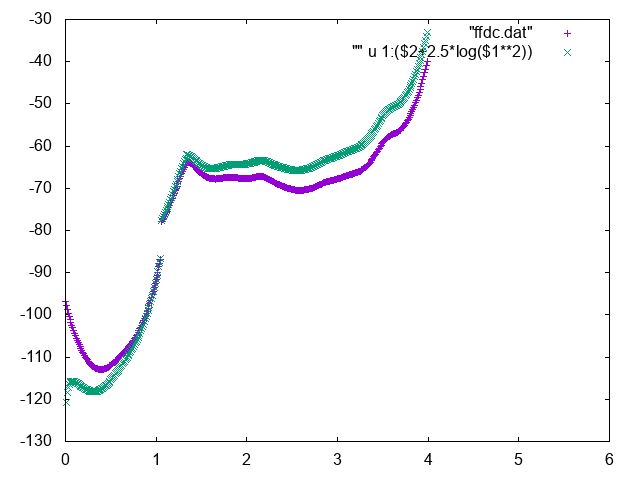
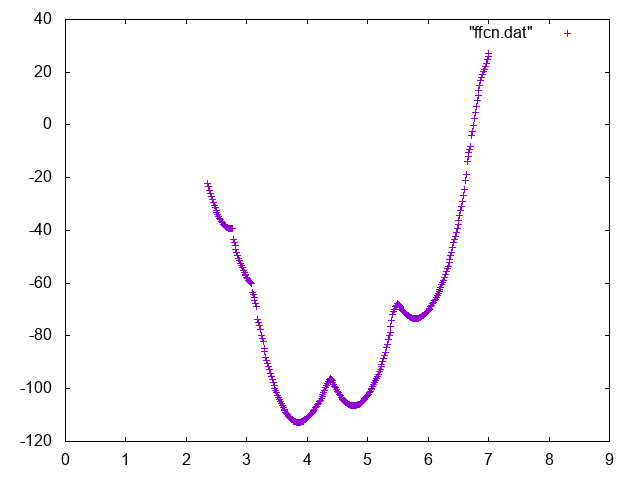
Also try to compute conditional free energies:
A possible way to do so you can use UPDATE_IF to extract portions of trajectory such that the Mg is bound or unbound.
Below you can find reference results
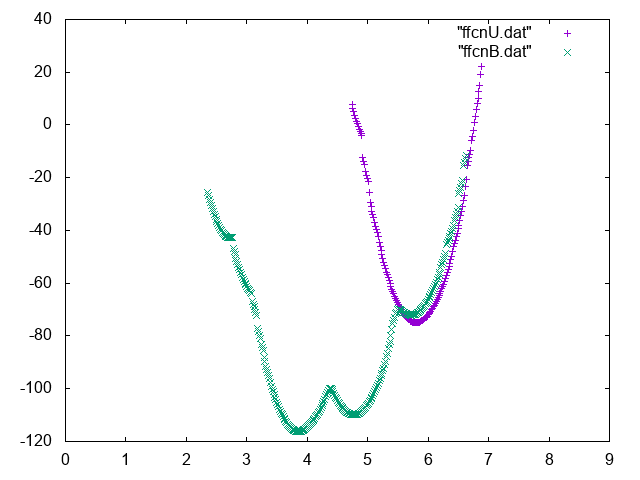
Now use the weights that you computed in the previous exercise to compute the absolute binding affinity of the Mg to the phosphate. In order to do so you should compute the relative probability of seeing the Mg bound to the phosphate and in the bulk region and normalize to 1 mol/M concentration.
For instance, if you define bulk the region between 1.5 and 2.5 nm, you should divide the weight of the unbound state by a factor \( \frac{4\pi}{3}(2.5^3-1.5^3)/V_{mol} \) where \(V_{mol}=1.66 nm^3\) is the volume corresponding to the inverse of 1 mol/L concentration. The absolute binding affinity is then defined as \(-k_BT \log \frac{w_B}{w_U} \). You should obtain a value of approximately 50.4 kj/mol
 1.8.17
1.8.17